
One hundred million silver bullets: Generative medicine vs. cancer

Two tumours
I never met my grandfather.
P. Sankaran died in January 1979, when my father was just 19, from cancer that metastasized to his liver, but very likely originated in his pancreas. He survived 6 excruciating months from his diagnosis, enduring the agony of his disease and the torture of chemotherapy, before passing on far too young at the age of 56.
Towards the end, my father, who loved him more than his own life, took to camping outside the godman Sathya Sai Baba’s ashram in Puttaparthi at dawn, hoping to meet the baba during his morning darshan and convince him to save his father in exchange for a lifetime of servitude. On my father’s return to Bombay, he brought my grandfather holy ash, and told him – pleaded with him – that it would be alright, that he would recover. P. Sankaran smiled gently, held up three fingers, and said nothing. Three days later, he was gone.
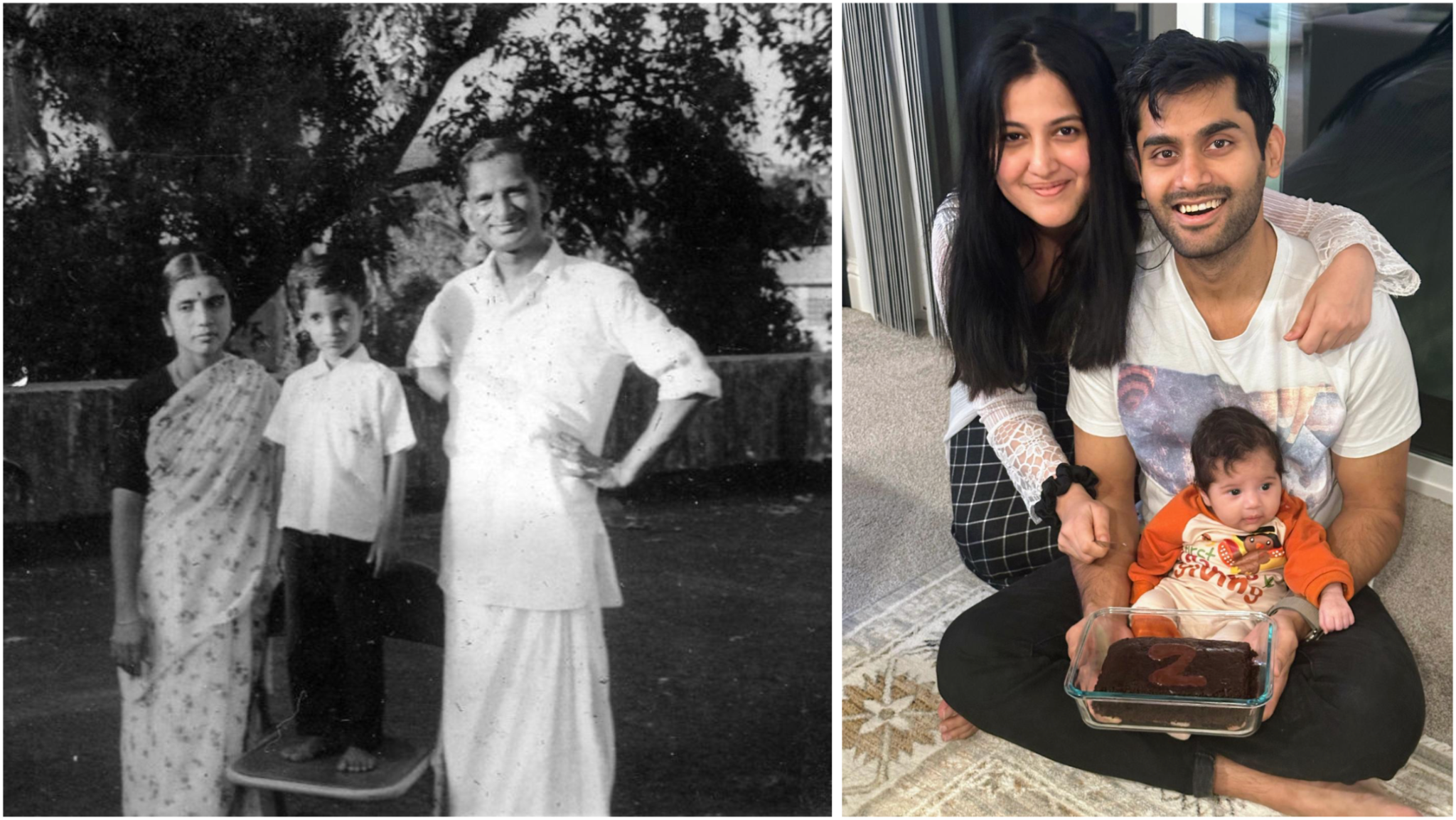
46 years later, in January 2025, Yash Bindal stepped into an emergency room in Palo Alto, California, on the other side of the world, with pain in his stomach and back. He’d just returned to his job at Meta from paternity leave after the birth of his daughter – his first child – only to find that he was persistently fatigued and nauseous, suddenly out of breath after a short run, very unusual for this fit 32-year old who’d played cricket for the Boston Gymkhana Club for half a decade in the not-too-distant past. He’d come around with these symptoms two weeks before and been dismissed with a diagnosis of constipation, but this time the persistence of his pain motivated the doctors to perform an ultrasound. There it was, clear as day – a mass in his liver.
Over the next few days, blood tests, CT & PET scans, and a biopsy at the Stanford University Medical Center confirmed the worst-case scenario – a 4 cm active tumour in his pancreas, which had spread to multiple lesions in his liver, and the shadow of micronodules in his lungs. This was advanced pancreatic cancer, already at Stage IV.

Nearly half a century after my grandfather breathed his last in a Bombay hospital, there are still no particularly good options for treating pancreatic cancer. The single most effective method remains the surgical removal of a substantial portion of the pancreas using what is known as the Whipple procedure, or, if the tumour is too large, complete surgical removal of the pancreas, but surgery of any kind is only viable in about 20% of cases, where the cancer is detected before it has spread further – most cases are diagnosed once the tumour has already ‘locally advanced’ to infiltrate nearby lymph nodes and major blood vessels, or metastasized to other organs such as the liver, as in my grandfather’s case, and in Yash’s.
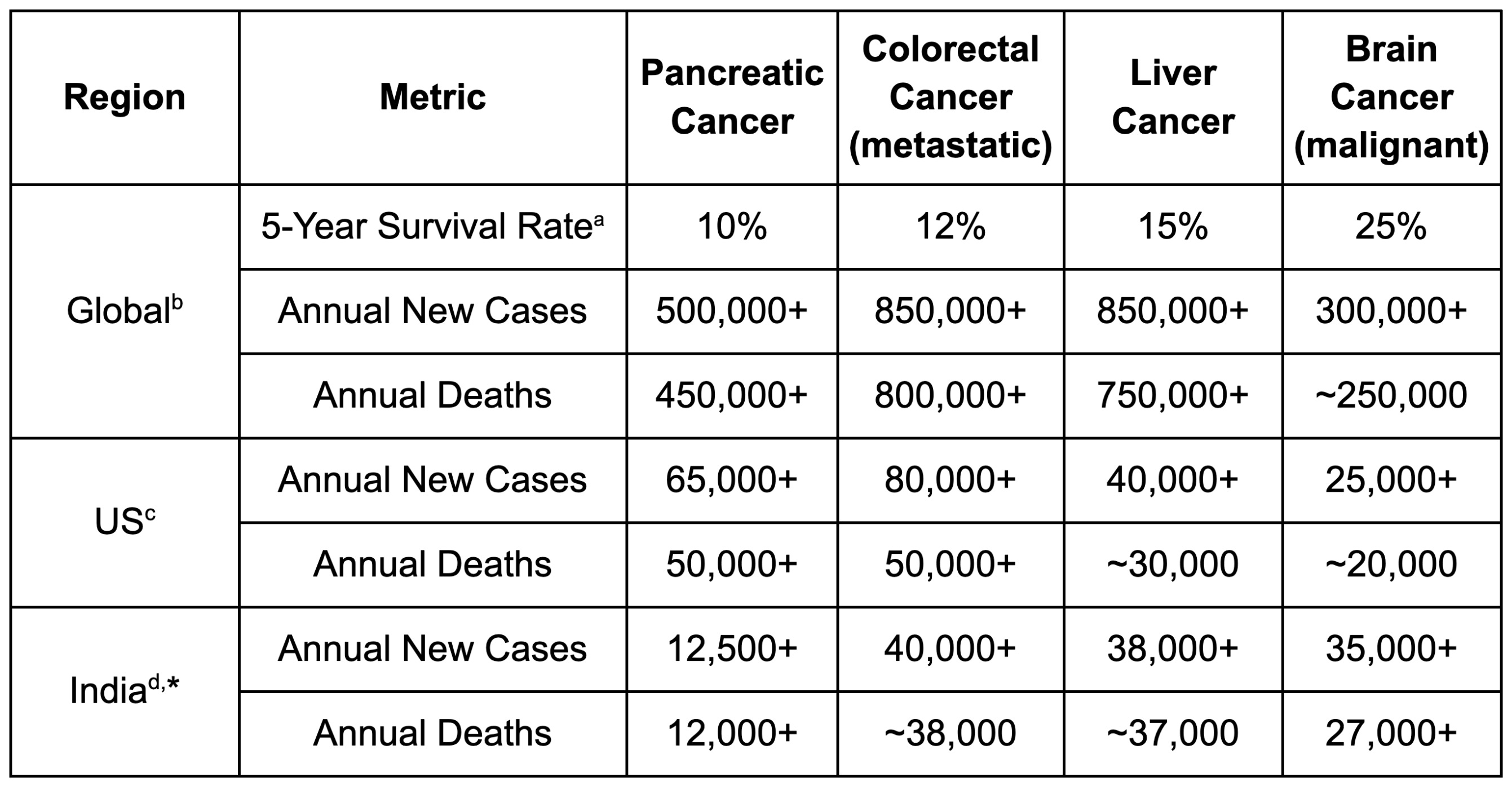
This lack of effective treatment has resulted in a 5-year survival rate for pancreatic ductal adenocarcinoma (PDAC), the subtype of the overwhelming majority of pancreatic cancer cases, of around 10%, even in wealthy countries such as the United States. In comparison, with access to modern standard-of-care therapies, the average 5-year survival rate across all cancers is over 50%.
As Yash and his wife Garima learned in the aftermath of his diagnosis, at Stage IV, with metastases to other organs, the 5-year survival rate is even lower – less than 3%.
Most patients are dead within a year of the cancer being first detected.
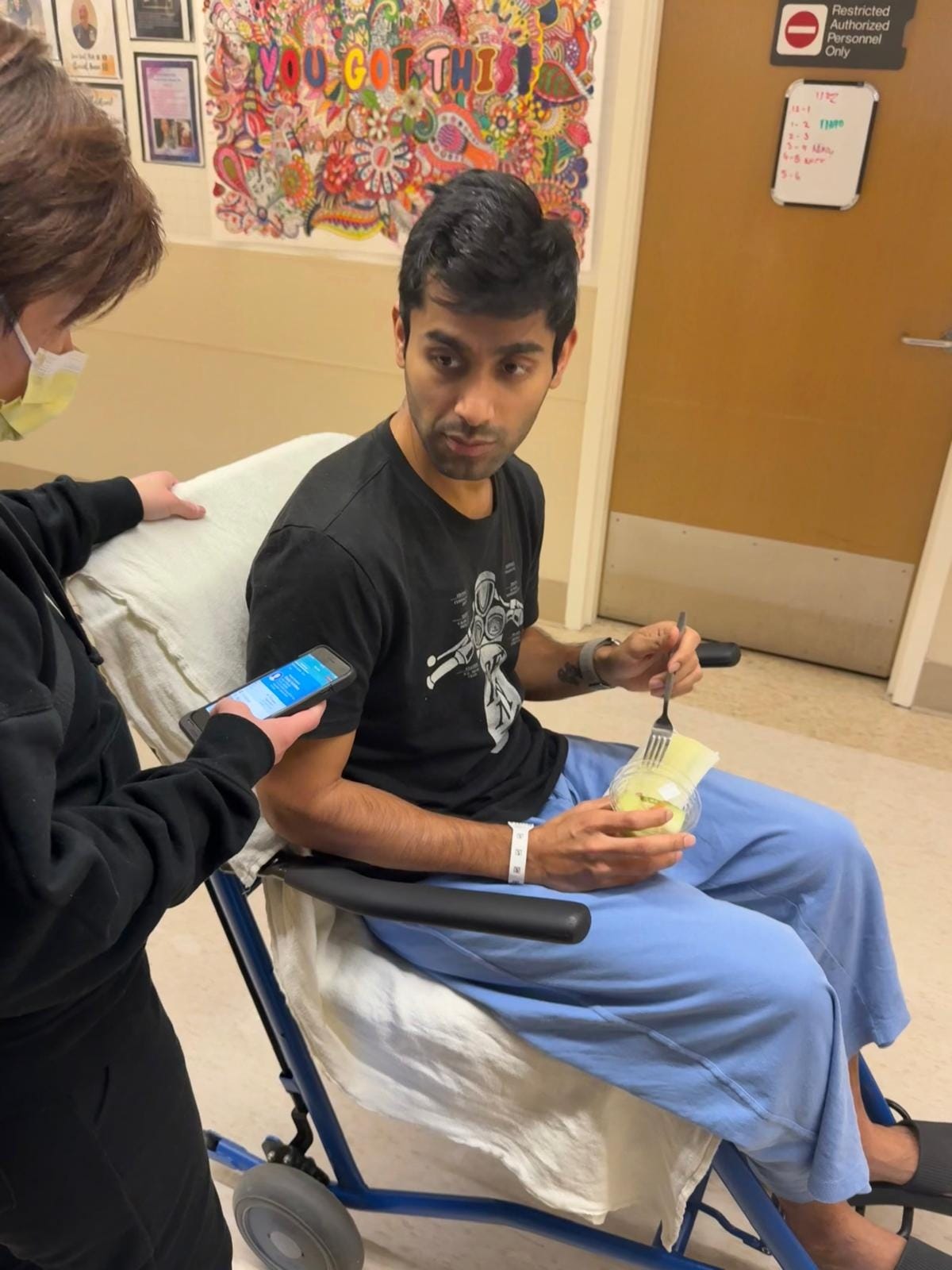
For most locally advanced and metastatic cases, chemotherapy regimens like NALIRIFOX, FOLFIRINOX, and Gemcitabine/Abraxane are the gold standard of care, but they are usually only able to extend patient survival by 6 months or less. These punishing multi-drug combinations often have severe side effects, leaving patients unable to taste the simple pleasures of everyday life in their last few thousand hours on earth. On the 17th of January, the day after his biopsy, Yash started FOLFIRINOX. The first cycle nearly killed him – severe anaemia set in, and he needed five blood transfusions to keep him going. My grandfather endured his own version of the same torture in 1978 – the names of the drugs have changed, but the pain has not.
The most exciting new chemical drug (‘small molecule’) developed against pancreatic cancer in decades, Revolution Medicines’ daraxonrasib, has been hailed as a potential game-changer in the treatment of this disease, with the just-released summary Phase III trial data demonstrating an increase in overall patient survival of only 6.5 months on top of existing chemotherapies. That excitement makes sense when you consider that this mere 6.5 extra months represents a doubling – a doubling! – of the life expectancy of these kinds of pretreated pancreatic cancer patients.
Each year, around 500,000 people are diagnosed with pancreatic cancer across the world, and over 450,000 people die from the disease, with the US alone accounting for more than 60,000 new cases and 50,000 deaths. These numbers appear to have more than doubled since 1990, and the number of pancreatic cancer patients under the age of 55 has been increasing on an even sharper trajectory, particularly in younger women.
An effective therapy for PDAC would not only save millions of lives across the world, but also spare millions more the pain my father suffered during and after his father’s untimely passing – the unbearable, indescribable pain of losing their fathers, mothers, brothers, and sisters in the blink of an eye to the ravages of this deadly disease.
And pancreatic cancer is not alone. Hepatocellular carcinoma, the most common form of liver cancer, also remains devastatingly lethal, with a 5-year survival rate under 20%. Many aggressive solid tumours still carry survival rates that would seem scandalous if we were not so accustomed to saying their names with clinical detachment.
Immunotherapies haven’t yet managed to cure cancer
You might wonder why cures for these cancers haven’t already emerged. After all, we live in a golden age of new cancer immunotherapies, a class of medicines which reprogram one’s own immune system to better fight tumours. Monoclonal antibodies, multi-specifics, cell therapies, cytokines, cancer vaccines – the variety is breathtaking, and the speed of new clinical trial initiation is dizzying, especially since the Chinese have entered the battlefield, all guns blazing. Every week, there is news that a different type of tumour may have fallen to the relentless onslaught of medical progress.
The flagship drug of the cancer immunotherapy revolution is Merck’s Keytruda, née pembrolizumab, an immune checkpoint inhibitor that blocks cancer cells from binding the PD-1 protein on T cells to trigger immunosuppression and prevent themselves from being killed. It made over $30 billion last year1, and has attracted a legion of competitors and imitators targeting the similar mechanisms that collectively made over $15 billion last year besides.
And yet, and yet, and yet… each of these medicines is able to help merely a fraction of a fraction of the cancer patients that are diagnosed each day across the world. Each of them has complex inclusion and exclusion criteria, sometimes supported by multiple bespoke diagnostic tests. Even immune checkpoint inhibitors like pembrolizumab only seem to benefit 20% of US cancer patients2. Before the arrival of these therapies, a patient with metastatic Non-Small Cell Lung Cancer (NSCLC) had a 5-8% chance of being alive in 5 years, a figure that the use of immune checkpoint inhibitors may have roughly doubled to 10-15%, but that’s still more than 5 out of every 6 patients not making it to the half-decade mark.
In the case of PDAC, immune checkpoint inhibitors such as Keytruda have shown almost no effect in multiple clinical trials, except in cases with rare genetic conditions like dMMR, which is present in only 1-2% of patients3. These past failures seem to have scared biotechs away from the disease – there are only a few immunotherapies targeting pancreatic cancer in the collective pipeline of the global pharmaceutical industry.
Like most pancreatic cancer patients, Yash wasn’t eligible for Keytruda, and unlike most pancreatic cancer patients, he wasn’t in the main target group for daraxonrasib – still unapproved – either. This drug, which is currently extending the life of the former US Senator from Nebraska (and fellow Yale alum) Ben Sasse, who was recently diagnosed with Stage IV pancreatic cancer, targets the RAS pathway, which is among the main drivers of the growth of up to 90% of pancreatic cancers. Yash is in the 10% with no RAS mutations in his tumours. The drug has very painful side effects – Mr. Sasse has been bleeding profusely from his face, which causes him pain that he describes as ‘nuclear’.

Sequencing of a tumour biopsy revealed that Yash’s cancer was likely driven by an NRG1 fusion mutation, in which the protein NRG1 fuses together with another protein to form a neoantigen which is not present on any healthy cells. NRG1 fusions are found in less than 1% of PDAC cases4 – Yash’s specific NRG1 fusion was with CDH1, a particularly rare partner even within this subset.
But timing was on his side. Just a month before he was diagnosed, a targeted therapy – zenocutuzumab, marketed by the Dutch biotech Merus as Bizengri – was approved precisely for cancers such as his. NRG1 fusions drive tumour growth by overactivating HER2-HER3 signalling – this antibody was designed to interrupt and block this interaction5. Thanks to the FOLFIRINOX, followed by Bizengri, Yash long surpassed the expectations of his dire prognosis. Nine months after his diagnosis, he was still playing cricket every weekend, still spending quality time with Garima and his daughter Maya, still going to work, still laughing at his own jokes.
Alas, mere precision did not produce a cure. Modern targeted cancer therapies are able to somewhat extend lifespan, but they are often unable to cure the disease itself – in many, if not most cases, while the medicine wins a battle or two, the cancer comes roaring back and eventually wins the war. For Yash & Garima, the unwelcome news came 6 months after he started treatment with Bizengri – his lung nodules had started to grow again. In the paradoxical language of oncology, he had ‘progressed’ beyond stasis into an uncertain future.
This sort of resurgence is common even when the cancer is caught early – recurrence of liver cancer after surgery to excise the tumours, which is generally only possible at Stage I and almost unheard of past Stage II, occurs in 70% of cases within 5 years6.
One of the few types of immunotherapy that has shown promising signs of longer-term efficacy against solid tumours in clinical trials is Chimeric Antigen Receptor T-cell therapy (CAR-T), where a patient’s T cells are extracted and externally engineered to express a binder that targets a specific tumour cell marker. The highest tested dose of the GCC-targeting GCC19CART, for example, seems to have added a year or more to the lives of patients with metastatic colorectal cancer for whom multiple other therapeutic options had already failed.
Unfortunately, CAR-Ts are extremely difficult to manufacture quickly – you have to take blood from the patient, get it to the manufacturing facility, separate the T cells, engineer them to express the binder using a viral vector, check that they are indeed expressing the binder, expand the population of cells expressing the binder, ensure that the cells haven’t been contaminated with any pathogens or toxic materials during this process, ship the cells back to where the patient is, and re-inject them into the patient. Before you do any of that, you have to evaluate whether the patient is a good fit for the CAR-T therapy and secure a production slot with the manufacturer. At its very fastest, this kind of autologous CAR-T takes just over a month end-to-end, but that can easily extend to 2 or even 3 months, by which time patients with aggressive solid cancers like PDAC may well be dead. Attempts to use pre-engineered T cells from other individuals which could be administered off-the-shelf – allogeneic CAR-T – have largely failed due to safety concerns.
In addition to the rare CDH1-NRG1 fusion mutation, Yash’s tumour overexpresses multiple known cancer antigens that are relatively common, but there are no approved CAR-T therapies that target any of them. The bespoke infrastructure and painstaking process development required to even bring therapies into clinical trials, much less manufacture them at scale, have contributed to the unfortunate reality that no CAR-T for any solid tumour target has yet been approved. Nearly a decade after the first CAR-T received FDA approval in 2017, all seven approved CAR-T therapies still collectively span only two targets, CD19 and BCMA on B cells. Manufacturing isn’t the only bottleneck; there are serious biological barriers in the tumour microenvironment (TME) as well, but if this type of therapy were simpler to develop and manufacture, there would be far more of them in the clinical pipeline.
What if we could direct a patient’s T cells towards the tumour without having to take them out and engineer them? In 2014, the FDA approved a new medicine that did just that – Amgen’s Blincyto (blinatumomab), the first T cell engager (TCE). It works by simultaneously binding to CD3 on T cells and CD19 on B cells, forming a bridge that pulls the two cell types into close proximity and triggers the T cells to kill the B cells. Like the very first CAR-T therapies, which also targeted CD19, Blincyto is intended to be deployed against cancers like B cell acute lymphoblastic leukemia (B-ALL), in which the cancer cells have replaced a substantial portion of the healthy blood cell precursors in the patient’s bone marrow by the time of diagnosis, choking off the production of normal blood cells.
Over the past few years, TCEs have become one of the hottest areas of medicine, with multiple billion-dollar deals announced each year – Gilead did one just a few weeks ago, buying Ouro Medicines for over $1.5 billion. In 2022, the FDA approved Immunocore’s Kimmtrak (tebentafusp), the first TCE with a solid tumour target – a peptide fragment of gp100 in patients with metastatic uveal melanoma, a cancer of the eye. Amgen’s Imdelltra (tarlatamab), a TCE targeting delta-like ligand 3 (DLL3), which is overexpressed in small cell lung cancer, was approved in 2024, bringing this approach to bear against one of the deadliest cancers known to man – one that carries a 5-year survival rate under 10%.
Most TCEs are, however, built around two or more antibody fragments linked together. Like with conventional monoclonal antibodies, you have to coax mammalian cells, often of the Chinese Hamster Ovary (CHO) variety, to produce your protein in giant bioreactors, which is often inherently finicky in ways that defy stabilization. However, thanks to decades of producing monoclonal antibodies at scale, biotechs have figured out how to rapidly develop GMP processes to produce them with high yield and efficiency.
How hard could this be?
But here’s the thing – a monoclonal antibody is a beautifully symmetric molecule, two identical heavy chains paired with two identical light chains, a design honed by hundreds of millions of years of evolution to be reliably assembled in mammalian cells. A TCE throws that symmetry out the window. You now need two different heavy chains and, in many formats, two different light chains, all expressed in the same cell, and you need them to pair up correctly – the right heavy chain with the right light chain on each arm, and the two different halves coming together as a heterodimer rather than defaulting to the energetically-easier homodimer of two identical halves. The cell, of course, doesn’t know which chain is which, and it certainly doesn’t care about your intentions – it’ll happily spit out every possible mispaired combination alongside the one you actually want, and some of these mispaired species are close enough in size and charge to the correct product that separating them is a purification nightmare.
Clever protein engineering tricks exist to steer the cells in the right direction – knobs-into-holes mutations that make the two heavy chain halves physically fit together better than two copies of the same half, CrossMAb domain swaps, common light chain designs – but none of these are perfect, and even the best-engineered formats tend to result in more aggregation of the fragments into clumps and produce lower yields than a nice, clean, symmetric monoclonal antibody.
Even once a TCE is designed and tested successfully at lab scale, adapting and optimizing it for a larger GMP run can take two years or more – this process involves developing often painstaking purification protocols and bespoke analytical methods to prove that what you’ve put in the vial is actually what you think it is. By contrast, the same process can take as little as eight months for a monoclonal antibody.
This difference may be part of the reason why a second FDA approval for a TCE only happened in 2022, over seven years after Blincyto. Focusing on manufacturability also limits the available design space – if you have an excellent binder that just doesn’t purify well, tough luck! TCEs, it turns out, are easier to manufacture than CAR-Ts, but not quite easy enough.
What if we could package the effects of CAR-T into an mRNA-based delivery mechanism?
At PopVax, we build Hydaptors – mRNA-encoded T cell engagers. mRNA encodes the instructions to make a protein, not the protein itself, which makes it far easier to manufacture. Unlike a therapeutic protein, which is produced inside living cells maintained in large-scale artificial cultures outside of the body, mRNA is produced in a test tube via in vitro transcription (IVT), an enzymatic, largely cell-free process that makes RNA directly from free nucleotides using a small amount of template DNA made in bacteria. This RNA is easier to purify, with much less variation in process and yield from sequence to sequence than from protein to protein. This mRNA is then encapsulated into a lipid nanoparticle (LNP), a fat bubble made from multiple lipid components, in order to allow it to enter into the patient’s cells, which then produce the TCE protein themselves, releasing it into the bloodstream to find its target.
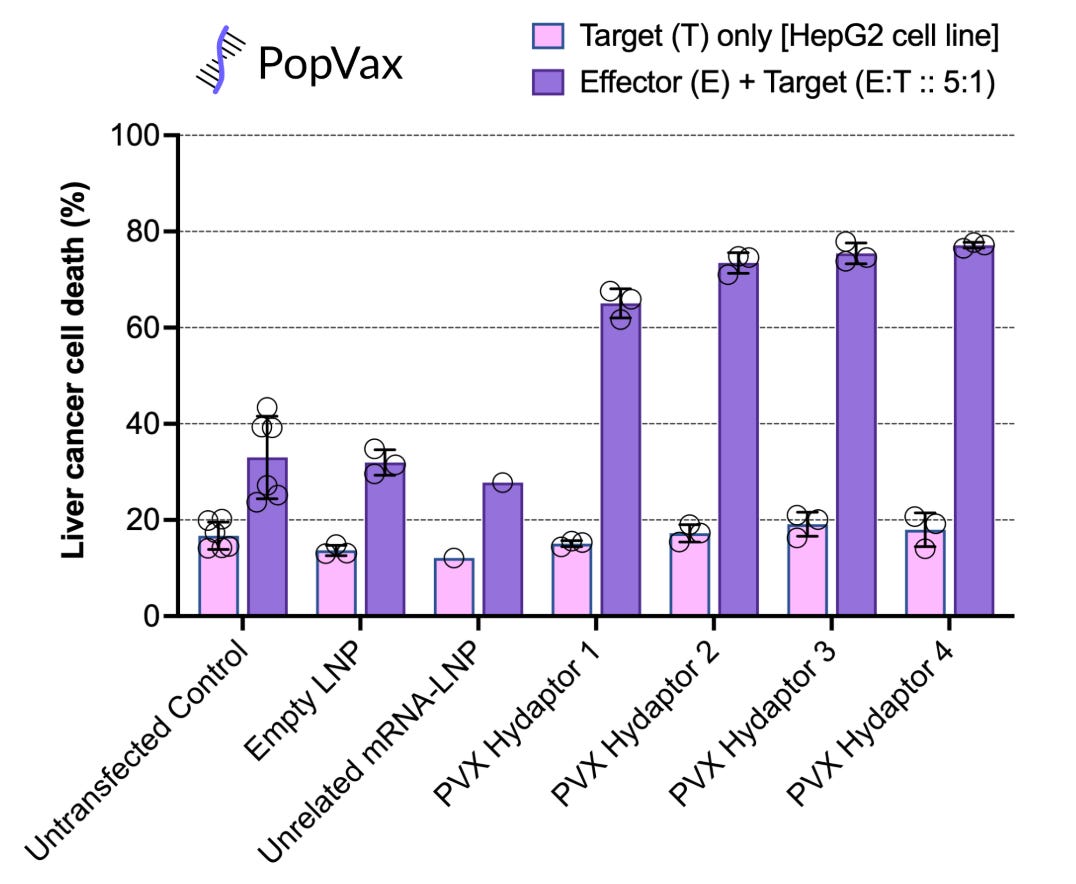
PopVax has designed and tested Hydaptors against Hepatocellular Carcinoma (HCC), the most common form of liver cancer, and found them to be highly effective at specifically killing liver cancer cells in a petri dish, while leaving other types of cells untouched.
Similar to the mRNA vaccines that proved to be the fastest to FDA approval during the COVID-19 pandemic, it has become fairly clear to us that using mRNA-LNP as the delivery mechanism makes these types of therapies faster to design, faster to manufacture, and faster to iterate on – a new mRNA therapy can be manufactured at GMP quality in weeks, not months or years. The Hydaptor modality eliminates the manufacturing bottleneck that has slowed CAR-T and TCE development over the past decade, and should help substantially accelerate the pace of bringing to clinic potent, targeted immunotherapies against cancers like Yash’s.
Even still, the best therapies of these kinds likely still won’t cure cancer. Over half of CAR-T patients, for example, will relapse within a year of treatment. There’s something fundamental about cancer that these therapies are failing to address.
Cancer is a many-headed hydra
Why is cancer so difficult an adversary to defeat? Cancer emerges when a cell mutates to multiply uncontrollably, overtaking, displacing, and starving cells around it. There isn’t one single pathway that makes this happen – the many, many possible combinations of causal mutations result in a dizzying array of manifestations with radically divergent characteristics and trajectories, including huge variation in what sorts of treatments are likely to be effective.
Consider Hepatocellular Carcinoma. HCC tumours usually have 50+ different aberrant mutations, several of which may substantially contribute to the manifestation and growth of the cancer. Different tumours of the same cancer type may, and in fact do, respond very differently to the same medicine, even if it is specifically targeted to that cancer type.
As my friend Abhishaike, the wise Owl who Posts, recently wrote, cancer has a surprising amount of detail. The field has spent decades understanding the ‘words’ of cancer – biomarkers like PD-L1 and HER2 – but now we face the challenge of understanding its grammar. As he puts it, ‘“HER2-positive” is a word. “HER2-positive, PD-L1-high, tumour-mutational-burden-high, tertiary-lymphoid-structure present, with exhausted CD8 niches” is a sentence’. When you squint at it, it becomes clear that cancer isn’t one single disease – it’s a collection of cellular growth disorders with somewhat similar symptoms and a shared set of progenitor mutations.
Today’s immunotherapies target one or at most a few pathways of cancer growth, or reprogram killer immune cells to go after a single marker expressed on the surface of cancer cells. The basis set of enabling mutations for any given tumour, however, typically includes alterations to genes like TP53 and BRCA1 which produce proteins that help repair DNA damage. As these natural tumour suppressors are rendered nonfunctional, and cancer cells divide faster and faster, the tumour inevitably picks up more and more mutations, reducing its dependence on any one pathway of growth, and providing evolutionary pathways to reduce the expression of the markers you might target, thus abrogating the efficacy of the administered immunotherapies over time.
In essence, a tumour is not a monolith, but a heterogenous population – when you hit a single target, you do not destroy the cancer, but rather inadvertently select for what remains. The fractal nature of the disease results in a combinatorial explosion, making the fragmentation of drug development to fight it seem inevitable. After all, if it isn’t one disease but millions of unique maladies, surely we’ll need to have thousands of companies developing tens of thousands of different therapeutics to combat each driving mutation – at today’s cost of at least $1 billion to get a drug to FDA approval, this would take tens of trillions of dollars. The world spends about $50 billion a year on cancer research – at this rate, will it take us more than 200 years to cure cancer?
Perhaps not. Cancer is a wily adversary – it takes many forms and wears many faces, shifting shapes in the shadows, poking and prodding at our immune defenses until it finds a weakness or three to metastasize through. It suppresses and exhausts immune cells in its vicinity, even recruiting them to its cause. Shining a light through the fog reveals the silhouette of a hydra that grows a new head somewhere different from the one you cut off the last time.
But if the disease can adapt, so too must the cure. We must fight fire with fire and create an immunotherapy as crafty and ruthless as cancer itself, less a single silver bullet than a swarm of drones attacking from all sides with all manner of explosives and ammunition, trapping the serpent in a surprise pincer movement.
What if we could attack multiple targets on each person’s cancer individually, boxing it in all at once?
What would it look like to hit not one, but many specific targets on a patient’s cancer all at once, with the intent to box in the disease’s evolutionary pathways and have a genuine shot at clearing it altogether? What if we could customize this for each patient, for each cancer, for each tumour?
The closest anyone has come to this are the clinical-stage personalized mRNA cancer vaccine programs developed by Moderna and BioNTech. They sequence individual tumours and encode 20-40 fragments of the mutated cancer progenitor proteins they find into mRNA, which they inject into the patient with the goal of priming T cells to kill cancer cells displaying these fragments on their surface.
Early results for some of these programs have been promising. Moderna’s mRNA-4157 (V940), for instance, reduced the risk of recurrence by 49%7 over three years in melanoma patients, and trials are underway to test the efficacy of these vaccines against non-small cell lung cancer and cervical cancer, among others. In a phase I trial of BioNTech’s BT-1228, a personalized vaccine against pancreatic cancer, none of the participants who mounted a strong immune response had a recurrence of their cancer within 18 months of administration, whereas the average time to recurrence for those who didn’t show a strong immune response was less than 12 months.
Unfortunately, these vaccines have three major problems.
First, the T cells that these vaccines intend to activate do not target whole proteins – instead, they recognize short peptide fragments from those proteins displayed on the cell surface. The molecular carriers that decide which peptide fragments get displayed, encoded in the Major Histocompatibility Complex (MHC), vary enormously across people, which means that a fragment of a neoantigen encoding a particular cancer mutation – the epitope – may show up on the cell surface in one person, but not in another, despite the underlying mutation being present in both. This means that even if a cancer vaccine generates a strong immune response against a particular neoantigen in a patient, it may have no effect because the neoantigen epitope the elicited T cells are targeting isn’t displayed on the surface of that patient’s cancer cells.
Second, even if a given epitope is correctly identified as displaying on a patient’s cancer cells, and is included in the vaccine, it may not be immunogenic enough to elicit a strong T cell response – most epitopes aren’t! In the BioNTech pancreatic cancer trial, in which they encoded as many as 20 neoantigen epitopes into each personalized vaccine, only an average of 1 in 10 epitopes was strong enough to trigger a specific T cell immune response against itself. In half the patients, not a single neoantigen successfully triggered a response.
Cancer vaccines try to attack all of the hydra’s heads at once, but to actually help clear tumours, a given neoantigen epitope in a vaccine has to be at the intersection of those displayed on the cell surface and those able to elicit a strong T cell immune response, two properties which are not necessarily correlated with each other. As a result, these vaccines end up acting like revolvers with at most a single live round loaded in the chamber – the vast majority of their shots end up being blanks.
Third, these trials have focused on patients with early-stage cancer, often those whose tumours were detected early and surgically removed – regardless of cancer type, these are the people who have the best chance of survival. Trials of personalized vaccines in late-stage cancers such as Yash’s haven’t (yet) yielded particularly positive results, which may be because of the indirect nature of the therapeutic effect – you’re relying on the patient’s body to produce an immune response, and the potency of that natural-like immune response has a hard limit that is perhaps too low to displace large, entrenched, or widely dispersed tumours.
It is much more effective to directly engineer T-cell behavior against specific cell populations, as effected by CAR-Ts, T cell engagers, and Hydaptors – these types of modalities are typically much more potent in clearing tumours.
What if you could effectively attack all of the relevant targets on a patient’s cancer at once, as the vaccines are trying to do, but with something as powerful as CAR-T? The problem is that unlike a cancer vaccine, where you have to merely select the right neoantigen epitopes to encode in your vaccine – which is hard enough! – for CAR-Ts and TCEs you need to actually design binders against these epitopes to point immune cells at their targets. It’s typically taken months, if not years, to design and refine these binders for use in a clinical trial – in the old paradigm, designing several of these after sequencing a patient’s tumour, and then engineering the patient’s cells to express them all at once, would certainly not be possible before patients die from diseases like PDAC and metastatic HCC.
Enter generative AI.
Potent personalized cancer immunotherapies are now possible with generative AI for protein design & the mRNA-LNP platform
PopVax commenced operations in late 2021, just a few months after the publication of the AlphaFold 2 paper, which revolutionized the prediction of protein structures from sequences. We’re now in 2026, and over those 4.5 years, generative AI for protein design progressed astoundingly quickly from being a theoretically interesting notion to very real and very useful. The 2024 Nobel Prize in Chemistry was awarded jointly to John Jumper & Demis Hassabis for AlphaFold, and to David Baker for his pioneering work on computational protein design over several decades, culminating in generative protein design models like ProteinMPNN and RFdiffusion, which use the same architecture as image & video generation models like Midjourney, Sora (RIP), and Veo.
We initially applied these approaches to design immunogens for broadly-protective vaccines. Design is nothing without validation and the means for translation into the real world, so in parallel we built out world-class integrated infrastructure equipped for molecular biology, immunology, chemistry, nucleic acid delivery, formulation, analytical R&D, and a versatile fast-turnaround GMP facility allowing for clinical dose production under one roof at our RNA Foundry. We’ve leveraged this full-stack approach to bring our first vaccine, built on our own computational design and mRNA-LNP platforms, to the brink of clinical trials – the Phase I will start in Australia in just a few months.

In mid-2025, we began expanding the scope of our research & development efforts to include cancer immunotherapy. This was a more natural fit than it may seem at first – we spent years designing and testing mRNA-encoded proteins intended to elicit very specific immune responses, and it turns out that requires the same set of methods you need to develop precision immunotherapies against cancer. Our work guiding generative models with internal immunological data proved to be extremely generalizable to this new domain.
Remember the Hydaptors – PopVax’s mRNA-encoded T cell engagers I described before? We de novo designed the binders that guide them to liver cancer cells using our AI-powered protein generation, ranking, and optimization pipeline, yielding a first-pass hit rate of 60% in a functional assay measuring their ability to drive immune cells to kill liver cancer cells in a petri dish, which is competitive with the best pipelines of this kind in the world. We tested 45, but we could have tested as few as 20 and still gotten potent Hydaptors. From design to in vitro validation, this whole process took about a month, of which an entire week was lost to customs delays in transit – a far cry from the pre-AI era of binder generation via animal immunization and phage display, where getting 10 hits might have taken a minimum of 4-6 months.
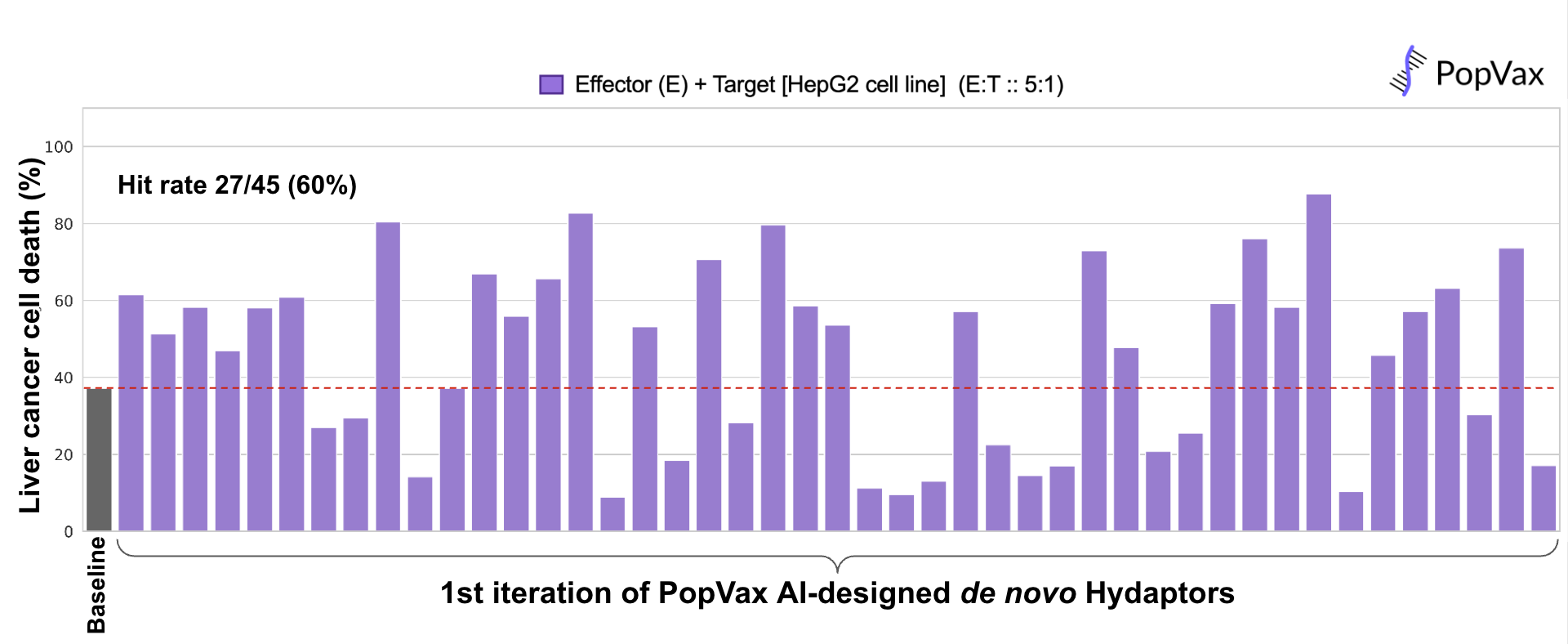
We now believe that AI models for protein design, guided with task-specific data and validated at high-throughput & low latency in the wet lab, combined with the speed & flexibility of the mRNA-LNP platform, have opened the door to the era of personalized generative medicine.
The ultimate cancer immunotherapy of the future will be less a single product than a generative AI-powered algorithmic process yielding a stack of precision-designed personalized therapeutics specific to each individual patient and their tumour, targeting multiple pathways and markers, boxing in the tumour and cutting off its evolutionary pathways all at once.
At PopVax, we’re making this future a reality. We’re combining the best aspects of both existing types of targeted cancer immunotherapy modalities – hitting multiple patient-specific cancer targets, as the mRNA cancer vaccines intend to, with the raw power of T cell redirection à la CAR-Ts and TCEs – to design, validate, and manufacture personalized Hydaptors designed to bind targets specific to a single patient’s cancer in just a couple of months from receiving tumour sequencing data, a timeline similar to today’s CAR-Ts. This is an entirely new class of therapy that couldn’t have been produced on reasonable timelines even 5 years ago, when protein design was in its infancy.
In fact, that future is already here, just unevenly distributed.
Sid & Amy & Andrew & Yash
Sid Sijbrandij was 43 years old when he was diagnosed with high-grade osteosarcoma – bone cancer – in 2022. After an aggressive course of chemotherapy, radiation, and surgery to remove a 6 cm tumour in his spine, his cancer went into remission – until 2024, when it came back with a vengeance. His options for conventional treatment had been exhausted, and he was too old to ever be considered for a clinical trial for his cancer, which typically occurs in much younger people. His doctors told him to begin putting his affairs in order and make the best of the time he had left.
At this point, he reached for a weapon that only a few thousand people in the world possess. Sid happens to be a billionaire, the co-founder of the software company GitLab, so he hired a team of experts and spent tens of millions of dollars sourcing and injecting experimental therapies for his cancer, including an oncolytic virus therapy intended to selectively infect and destroy his cancer cells, as well as personalized cancer vaccines designed to prod his immune system into eating away his tumour. The therapy that really seemed to do the trick was a radioligand therapy targeted at FAPα, found on cancer-associated fibroblasts (CAFs) that secrete proteins which create a shield around solid tumours, preventing immune cells and medicines from penetrating. This experimental therapy, manufactured and administered in a private clinic in Germany, wasn’t specifically built for him, but his TME was particularly rich in these CAFs, making him a prime candidate to receive it. It shrunk his tumour enough to allow him to undergo a second surgery to remove the rest.
Sid’s armoury of experimental therapy seems to have tipped the scales against osteosarcoma – today, his cancer is no longer detectable, two years after it seemed all hope was lost. This battle may be won, but he’s still fighting the war – he’s begun to fund next-generation logic-gated CAR-T therapies, as well as start up a whole bunch of companies to further develop the technologies that have helped him. In effect, he’s running a self-funded R&D program funneling therapies into a rolling series of human trials with an n of 1. His particular path, however, may be a bit harder to replicate without the many millions of dollars at his disposal. Nevertheless, Sid’s approach has already inspired people like Andrew Rodriguez, a certified non-billionaire whose girlfriend Amy was diagnosed with a fast-growing pituitary tumour at the base of her brain, to begin exploring experimental treatments and personalized therapeutics to cure her – he even wrote a widely-read article to enlist the wider world in this quest.
I met Sid over breakfast in Prospera, the libertarian charter city in Honduras, a few weeks ago, then heard him speak to an adoring crowd of biohackers in the surprisingly cozy Infinita Dome. I’d already spoken to Jacob Stern, one of Sid’s key team members, and read about Sid’s story in Elliot Hershberg’s excellent piece about him, but hearing Sid tell his story in person that day was electrifying and deeply inspiring. One thing he said stood out – a doctor friend of his, aghast at the wide variety of treatments that Sid was taking in parallel, told him
“Sid, if you take all of these therapies at once, how will we know what cured you?”
Something is deeply broken in the world of cancer care if stacking treatments against multiple targets, the approach most likely to provide a durable cure to the disease, is considered taboo.
The next day, I flew to the US with my colleague Darshit Mehta, lately of Ginkgo Bioworks and now back in India running programs at PopVax. We stopped over in North Carolina to meet with the oracle of open source cancer vaccines, Alex Rubinsteyn, whose OpenVax pipeline underlies much of the translational work in the field of personalized immunotherapy.
Then we flew to San Francisco to meet Yash.
Over a year since his diagnosis, and six months since progressing on the targeted therapy Bizengri, Yash is not only still alive – he is, frankly, still much fitter and better looking than me. He’s been on the Gemcitabine/Abraxane chemotherapy regimen combined with the tyrosine kinase inhibitor afatinib, which causes him serious digestive issues and severe pain, but he’s somehow still playing cricket every week, and has even found the time to illicitly eat jhol momos late at night with visiting biotechnologists at a hole-in-the-wall Nepali restaurant in San Mateo.
Yash and his tireless wife Garima have taken a leaf out of Sid’s book and taken control of their own cancer care. They swap notes in a Facebook group with other cancer patients with rare NRG1 fusion mutations, have insisted on much more frequent imaging of his tumours than his doctors were previously suggesting, and have even explored relocating to China to participate in a clinical trial for a targeted therapy there.
As Darshit and I sat across from Yash that night, scarfing down our baingan masala and dal chawal after a few days of Honduran beans and rice, we hatched a plan with him.
The CDH1-NRG1 fusion that he has is a rare surface-anchored neoantigen – it isn’t present in any healthy cells, but it is on the surface independent of the complex MHC display mechanism. We can be quite certain that it’s there, and that we can target it with an AI-designed Hydaptor.
That’s what we’ve resolved to do. Over the next 6 months, we will make Yash at least one, and maybe several personalized Hydaptor immunotherapies, de novo AI-designed generative medicines that we’ve just begun designing this week. We will test, validate, optimize, and manufacture these Hydaptors at our integrated RNA Foundry in Hyderabad, which has already been successfully inspected by Indian regulators for the GMP production of test lots of our broadly-protective mRNA vaccine against COVID-19.
We will then work with an expert clinical team to administer the therapy and monitor his safety. Finally, we will assess whether the therapy is able to substantially shrink his tumours, which is our short-term measure of success.
Our long term measure of success, of course, is Yash’s survival. We want him to live to see his daughter Maya go to school, grow up, find her calling, get married, and have children of her own if she so chooses; to age gracefully into old age with his beloved Garima; and if he does die far into the future, we don’t want it to be from pancreatic cancer.

We don’t know, or even expect, that the Hydaptor we build for Yash will be able to accomplish that – even a few more years without progression, without tumour growth, without further metastases, would constitute a major breakthrough. This probably won’t be the silver bullet for his cancer.
So we’ll build another one for him. And another after that, perhaps even 5 at a time. There are 20 million cancer cases diagnosed each year. If we hit each one with at least 5 personalized generative medicines, that’s 100 million silver bullets – a lot of heavy metal into the many heads of the hydra that is cancer.
And we’ve already begun.
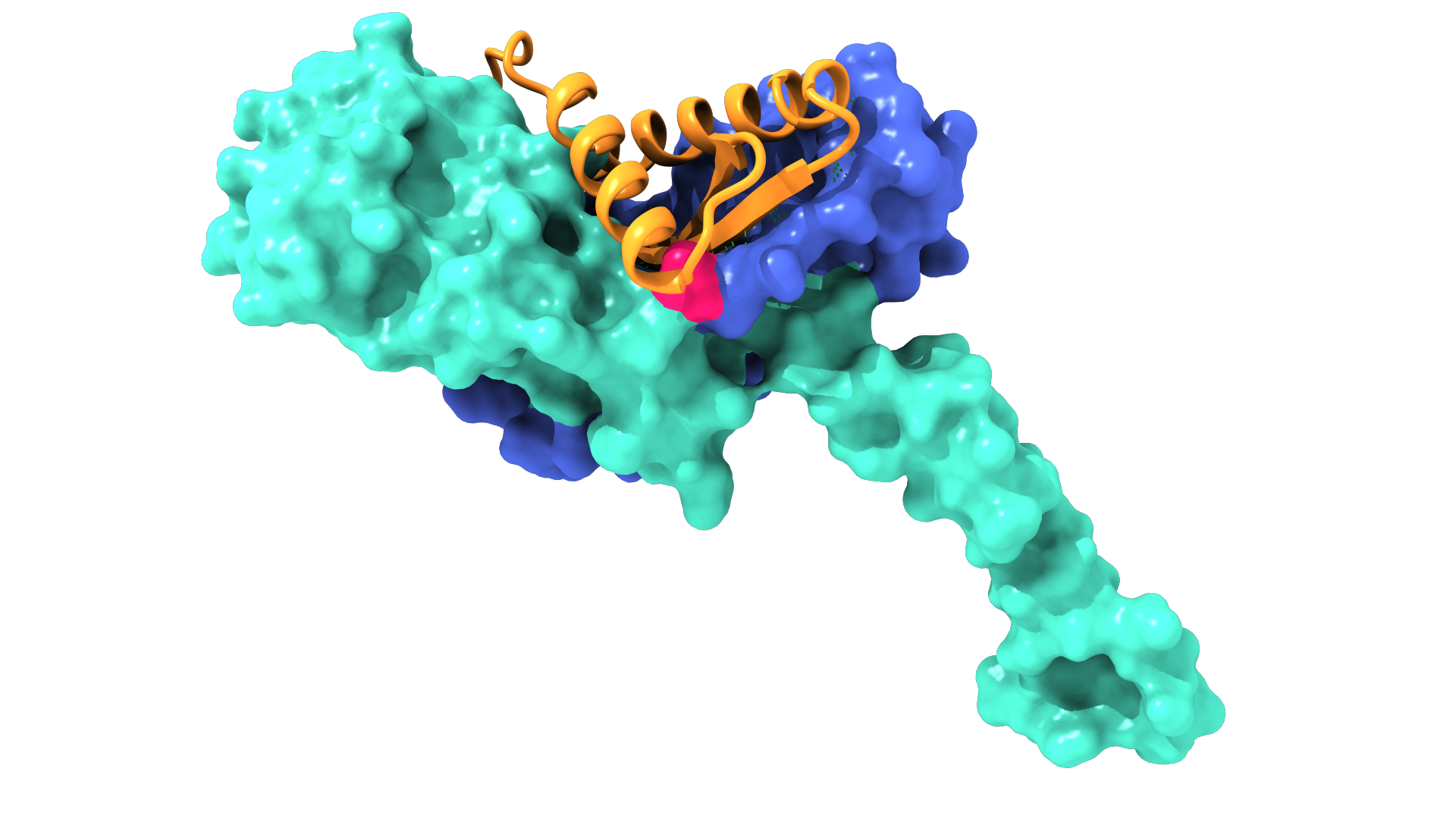
Making personalized Hydaptors safe, specific, and stackable
This won’t be easy. These personalized Hydaptors must be safe – most patients who take the current generation of targeted immunotherapies such as CAR-Ts and TCEs suffer Cytokine Release Syndrome or similar immunological side effects, where the immune response produced is so overwhelming that it becomes toxic to the body. This can both reduce the maximum possible dosage, forcing underdosing, and harm or even kill the patient. Dose too much, and the patient may be severely harmed or even killed by CRS; dose too little, and the cancer wins.
These Hydaptors must also be exquisitely specific – they must only target the neoantigen epitopes we want to hit, not bind with similar epitopes displayed on healthy cells and kill them by accident. We don’t want the cure to severely injure the patient – part of the problem with existing cancer therapies like chemo is the painful off-target effects. Take doxorubicin, one of the more common chemotherapies – as Abhi recently wrote in Owl Posting, this drug is nicknamed the ‘Red Devil’ because of the charming fact that it causes clinical heart failure in between 5% and 26% of patients, some of whom have died during the infusion itself! The Hydaptors must be bullets, not bombs.
If we achieve safety and specificity, we can make these Hydaptors stackable – we should be able to administer 2 or 5 or even 10 of them at once, all attacking different targets, at the same time. Combining the current generation of CAR-Ts, TCEs, and other targeted immunotherapies often kills the patient, because the collective toxicity to critical organs like the brain, heart, liver, and kidneys, both from off-target binding and CRS-like immunological inflammation, can kill the patient much faster than the cancer would. There is already evidence that sequentially administering immune checkpoint inhibitors like Keytruda and a CAR-T therapy may cause substantially more CRS and even neurotoxicity than either alone.
We can’t, of course, sacrifice functional efficacy at the altar of safety. Even today’s most potent CAR-Ts and TCEs struggle with clearing solid tumours, especially in pancreatic, liver, and colorectal cancers. These tumours are often surrounded by a protective wall of non-cancerous cells and fibrous proteins meant to isolate the tumour – to kill the tumour, we have to breach these walls to get immune cells and the immunotherapy into the tumour in the first place, which requires targeting the cells that create these barriers, like the FAPα-expressing CAFs that Sid’s radioligand therapy attacked. The difficulties don’t stop there – tumours often cultivate an immunologically ‘cold’ environment that can deactivate, exhaust, and deplete the killer T cells and other immune cells that do manage to enter, inhibiting the effectiveness of immunotherapies – we have to ‘warm up’ this environment, reprogramming and reactivating immune cells to allow immunotherapies to be effective.
A key insight that drives our vaccine work at PopVax is that binding between a designed immunogen and an antibody, and the ability of that immunogen to functionally elicit that antibody in vivo, are very much not the same thing – attempts to treat binding and function as interchangeable have resulted in decades of spectacular failure in HIV vaccinology despite billions of dollars of investment. This is also true for cancer immunotherapies – though high-affinity binding to the chosen target is a key part of making a successful Hydaptor, properties like functionality, specificity, and immune toxicity cannot be designed or even predicted conditioning on strength of binding alone.
We combine the power of generative AI for protein design with relentless empiricism – massively scaling innovative assays that directly link millions of unique protein designs to disease-specific functional readouts, all built and executed at high-throughput at our RNA Foundry, to train and guide generative models to produce safe, specific, stackable, and effective Hydaptors. We’re doing this in a personalized fashion for the first time for Yash, and we’re being conservative with our timeline. As we do this more and more, our models will get better and better, especially as we validate against clinical data, while our experimental validation & manufacturing workflows get faster and faster. What takes 6 months today will take just a couple of months next year, and just a few weeks not too long after that, which is the speed that truly personalized medicine demands, and we intend to achieve without trading one kind of harm for another.

Armed with the therapies we create with these methods, we aim to do nothing less than transform cancer treatment in the same way antiretrovirals transformed HIV treatment – ushering in a future in which cancer is not a certain, dreaded killer, but at worst a manageable problem whose patients continue to live long, productive lives. From that basecamp, we can then begin the climb to the summit of a true cure.
Let me digress slightly here to say a few things about the mRNA platform we’ve built the Hydaptors on. mRNA vaccines do have their disadvantages when used against infectious diseases. They are often more reactogenic than conventional vaccines, resulting in more immediate side effects like fever, muscle pain, and fatigue. More seriously, in a very small percentage of cases, they’ve been linked to myocarditis. PopVax and many others in the field are working tirelessly to build new RNA platforms that substantially reduce and eventually eliminate these adverse effects.
RNA does, however, seem to be the perfect platform for cancer immunotherapy. Recombinant peptide-based cancer vaccines, with the proteins expressed in cell culture and then mixed with adjuvants to administer, had been tried for decades and basically all failed. mRNA cancer vaccines were immediately much, much better at eliciting T cell response – it’s no coincidence that all the major personalized cancer vaccine programs that are in late-stage trials are RNA-based. Some intrinsic property of mRNA-LNPs seems to be able to cause beneficial immune stimulation against cancer cells, and perhaps warm up cold tumour microenvironments – indeed, a retrospective study9 at the MD Anderson Cancer Center reported that patients who received mRNA COVID-19 vaccines during treatment with immune checkpoint inhibitors like Keytruda had almost double the 3-year survival rate compared with those that received non-mRNA vaccines or no vaccine at all. mRNA-LNPs may also have an easier time entering solid tumour TMEs than CAR-Ts or traditional TCEs, allowing them to reprogram the TME from the inside.
With our full-stack approach to biology, PopVax integrates the entire therapeutic development pipeline, from generative AI for design to high-throughput wet lab testing to manufacturing, under one roof at our RNA Foundry in Hyderabad – this makes us one of the few companies in the world with the ability to deliver on the promise of personalized generative medicine from concept to clinic. Our in-house GMP production facility sits just through the glass from our R&D lab at the RNA Foundry, allowing us to seamlessly translate promising designs from lab bench to clinical-grade batch in weeks, not years, enabled by the inherent adaptability and efficiency of our mRNA-LNP platform.

The regulatory waters are parting to open a path for personalized generative medicine
Just a few years ago, there was no regulatory pathway to get personalized generative Hydaptors approved for general use. No major pharmaceutical regulator has yet approved a product where the sequence and structure is personalized for each patient. Autologous CAR-Ts come the closest, since they involve engineering a patient’s own cells, but the therapeutic sequences engineered into the cells are the same.
The good news for us, and for all cancer patients, is that as personalized mRNA cancer vaccines roll towards approval, regulators have had to come to grips with the problem of how to license them, and new pathways have begun to emerge.
For a long time, the US FDA’s little-known expanded access program has allowed for the administration of experimental therapy to single patients. Kyle Muldoon Jr., better known as Baby KJ, has become a particularly celebrated beneficiary of this of late – a scientific team led by Kiran Musunuru at UPenn and Rebecca Ahrens-Nicklas at the Children’s Hospital of Philadelphia developed a tailor-made gene editing therapeutic to treat KJ’s neonatal-onset CPS1 deficiency, a rare, generally fatal disorder that made him unable to metabolize any proteins. They administered this medicine last year, just 7 months after KJ was born, and he seems to be on the road to recovery.
Sid has also used this pathway extensively to receive experimental and personalized therapies, at least 3 times starting in 2023, and has received approvals within 72 hours. He’s on record saying “the FDA wants me to live!”. The UK, PopVax’s new second home, has a similar regime for the administration of named-patient therapeutics under their “specials” pathway, which conjures the delightful image of ordering a generative medicine as you would an esoteric food from a well-dressed butler named Jeeves.
While this mechanism enables well-resourced individuals like Sid and experienced research teams to administer personalized therapies, it is still a substantial bureaucratic exercise, and remains out of reach for the vast majority of patients. This mechanism also just doesn’t scale – if millions of patients were to regularly file these on their own, they might flood the FDA and clog up the agency’s capacity to review them in a timely manner. Until very recently, there was no mechanism for this type of personalized therapy to proceed from single-patient administrations towards approval other than standard clinical trials, which may not be feasible for the rare diseases that would benefit most from such personalization.
Just a few months ago, the FDA announced a new regulatory track that might bridge this gap – the plausible mechanism pathway, which allows for the rapid approval of highly-personalized therapeutic platforms for rare diseases after demonstrated successes in a few patients. In a piece in the New England Journal of Medicine (NEJM) co-written by FDA Commissioner Marty Makary, the agency laid out a few key criteria for eligibility for this pathway10:
There should be a specific genetic, cellular, or molecular abnormality, typically a mutation, which has clear connection to the onset of the disease being treated
The therapy should directly target the underlying or proximate molecular abnormality
There should be a well-characterized natural history of the disease in the untreated population – this pathway particularly applies to diseases that are nearly always fatal or deeply debilitating
There needs to be some method of confirming that the target was successfully hit, as well as some way to demonstrate improvement in clinical outcomes
While this pathway was originally designed with a particular focus of gene editing – the NEJM piece specifically references Baby KJ – Yash and the Hydaptor therapy we’re creating for him seem to fit all of these criteria. His rare CDH1-NRG1 fusion mutation, present in under 1% of pancreatic cancer patients, drives the growth of his tumours via a well-understood HER3-mediated mechanism, and he doesn’t have a RAS mutation, so that fusion is likely the main driver. Our Hydaptors will specifically target cells with the CDH1-NRG1 fusion mutations, as well as other known cancer-driving mutations he has, with the intention of eliminating the cancer at its root. We will track progress based on the extent to which we are able to melt away his tumours, as well as reduce their metabolic activity, and if he lives for even a year longer because of this therapy, he will clearly be demonstrating a substantially improved clinical outcome relative to other patients with pancreatic cancers similar to his.
This pathway provides us the running track for our regulatory sprint. To quote the NEJM piece,
“once a manufacturer has demonstrated success with several consecutive patients with different bespoke therapies, the FDA will move toward granting marketing authorization for the product.”
We will start with finding a small group of individual patients who each fulfill these criteria and have relatively rare cancers, for which conventional clinical trials would be difficult, demonstrate success with hopefully many of them, and then move towards approval for our platform as a whole, starting with rare terminal cancers for which approved targeted immunotherapies are few and far between.
We are quite hopeful that we can deliver substantial benefit to patient #1 – Yash – and we will certainly get better at this each time we do it, fine-tuning our models, optimizing our processes, and refining our deployment approach. Even if this specific plausible mechanism pathway isn’t adjudged by FDA to be compatible with what we’re doing, we are confident that we can work with them and other regulators to make similar pathways available for rare cancers.
We believe there is a path to making personalized Hydaptor therapies available to tens of thousands of patients commercially over the next few years, and millions over the next half-decade. Over the next decade, as enabling frameworks like the UK MHRA’s draft guidelines on personalized mRNA cancer immunotherapies are finalized and come into effect, we want to make this type of generative medicine available to all cancer patients across the world11.
We’re coming for the emperor of all maladies, and we don’t intend to miss. We won’t rest till the job is done.
We’re asking for your help to win this battle, and then win the war
Our battle with Yash’s cancer, and the war against cancer writ large, can’t possibly be won alone. The personalized generative medicines we’re developing are part of a broader feedback loop between expert medical practitioners, precision diagnostics, and parallel therapies. We want to work with whoever has the best skills and tools to give Yash the best chance of living a long and healthy life, with the added benefit of helping test, optimize, and validate the use of a suite of medical technologies in this context for the benefit of the many cancer patients who will follow in his footsteps.
So, taking inspiration from Sid, Andrew, and Amy, we’re asking the world for help. Here’s what we need most:
A doctor in the US or India who is able to perform a biopsy and give Yash custody of his own sample for organoid establishment and transcriptomics
The most accurate in vitro model to test our Hydaptors would be a patient-derived organoid (PDO) grown from Yash’s own tumour cells. There are now protocols that may allow us to make organoids of this kind from as little as 10-20 mg of tissue, which we believe will yield millions of cells and can be obtained from a single core needle biopsy with relatively low risk.
We seek an experienced interventional radiologist, hepatobiliary oncologist, or surgeon with expertise in pancreatic cancer who can consult with us and potentially perform a core needle biopsy of Yash’s liver metastasis. Following collection, the biopsy sample would need to be transferred to Yash and his family under storage conditions specified by the PopVax team for sequencing and patient-derived organoid establishment.
If this is you, or you can offer an introduction to someone who can do this, email us at cancer [at] popvax [dot] com.
Single-cell DNA sequencing (scDNA-seq) and transcriptomics (scRNA-seq), as well as long-read whole-genome sequencing (WGS), with both raw reads and analysis provided for all
We need a very, very precise molecular picture of Yash’s tumour. Yash has already undergone several biopsies, and some of his tumours have already been sequenced. We don’t, however, have access to the raw reads from those sequencing runs. Once Yash obtains a new biopsy of one of his tumours, we seek a partner who would be able to single-cell sequence Yash’s tumour (DNA & RNA, separately) and provide the raw reads, as well as ideally give us their own analysis of potential targets worth trying to hit, both pMHC and otherwise. We also need a provider to perform long-read WGS and provide the raw reads as well as their analysis if possible.
If this is you, your research lab, or your company, email cancer [at] popvax [dot] com.
An oncologist with experience in administering and monitoring CAR-T or T cell engager therapy who would be willing to consider serving as PI for an n=1 study under the FDA expanded access pathway, the UK specials pathway, or the Australian Clinical Trial Notification (CTN) pathway
Once we have designed a personalized Hydaptor and tested it experimentally in our lab, and if Yash and the PopVax team are confident that it is both likely to be safe and has a strong chance of efficacy based on that data, we will need a physician willing to review the data and consider serving as PI for a single-patient study, likely in the US, the UK, or Australia. In particular, this will need to be someone who has experience monitoring CAR-T or T cell engager therapy, and dealing with potential immunotherapy-specific complications like Cytokine Release Syndrome (CRS) and neurotoxicity. Ideally, we will engage with whoever this is going to be early on, and get their advice throughout the development of the Hydaptor, including the design of experiments to validate safety and efficacy, as well as the clinical plan.
These are not novel regulatory pathways – they have been walked before by many others, and we would particularly benefit from working with doctors who have already been part of these sorts of n=1 deployments in the past, but that isn’t an absolute necessity.
If this is you, email us at cancer [at] popvax [dot] com.
Funding for Yash’s Hydaptor
The sprint we’re describing – biopsy, sequencing, design, high-throughput testing, administration, monitoring – is resource-intensive, and none of these steps can be skipped. We don’t plan to charge Yash and his family for our part in this, but we’re an early-stage company in India, and not as well-funded as our American biotech peers.
Since this is our first time executing this end-to-end pipeline, and each stage depends on the one before it, delays at any step can compound. With dedicated funding for Yash’s Hydaptor, we would be able to accelerate the timeline to injection without compromising safety by comprehensively testing more designs for functionality and potential toxicity in parallel.
If there are high net-worth individuals, science funding organizations, or cancer foundations willing to help cover the cost of developing and manufacturing Yash’s Hydaptor, as well as potential costs of administration and monitoring not covered by insurance, we would love to have a conversation.
If this is you or your organization, email us at cancer [at] popvax [dot] com.
(Bonus) Immunopeptidomics on a tiny number of cells (20-100k)
Immunopeptidomics pipelines to identify pMHCs displayed on cancer cells typically require many millions of cells, which we won’t have to spare. We might, however, be able to spare 20,000-100,000 cells if all goes well. We’re interested in hearing from anyone with an immunopeptidomics protocol that they are confident will work on such a small number of cells.
If this is you, email us at cancer [at] popvax [dot] com.

100 million silver bullets
20 million people diagnosed with cancer every year
x 5 personalized generative Hydaptor therapies each
= 100 million silver bullets PopVax must create each year
We’re building the first bullet to load into our gun and take into our first battle with Yash’s cancer. Next, we’ll scale up the RNA Foundry to do this for thousands of patients under single-patient pathways. Once that’s proved out, and our models & methods have been optimized by clinical data, we’ll work with regulators to run the trials necessary to bring this to millions of people. Then we’ll build our 100 million bullets and fire them all at once into the many heads of the hydra – and if it falls over, dead, we’ll win the war.
Come help us build the foundations of a cancer-free future! Apply at popvax.com/jobs, or email work [at] popvax [dot] com.
I’m Soham Sankaran, the founder & CEO of PopVax. Feel free to email soham [at] popvax [dot] com if you’re interested in discussing personalized generative medicine. I wrote this essay in honour of my paternal grandfather, P. Sankaran – we never met, but I think we would have gotten along.
I couldn’t have written this without Yash and Garima’s trust and support, for which I am deeply grateful.
You can find me on Twitter/X @sohamsankaran. You can also follow PopVax on Twitter/X, Bluesky, LinkedIn, and YouTube, or email us at hello [at] popvax [dot] com.
At PopVax, Samarth Jajoo, our head of special projects, Maunish Barvalia, our Chief Science Officer, Sahaj Sankaran, our other board member, and Dhruv Arora, my chief of staff, all made substantial contributions to the writing of this essay.
